Estimation of multidimensional site frequency spectra from low-coverage sequencing data
A faster and more efficient implementation
It was a great pleasure to have collaborated with Thorfinn Korneliussen, head of the ANGSD team, on a new implementation for estimating the site frequency spectrum (SFS) from sequencing data. The manuscript is accessible here and the implementation is available in the latest version of ANGSD.
In this study, with joint first co-authors Alex Mas-Sandoval and Nate Pope, we developed a new
The SFS is one of the most important features in population genetic data, as it summarizes the distribution of allele frequencies. As such, it is widely used to infer signals of natural selection and/or demographic events. However, as previously demonstrated, estimates of SFS from high-throughput low-coverage sequencing data may be biased if the statistical uncertainty in genotype calling is not properly addressed.
In this study, with joint first co-authors Alex Mas-Sandoval and Nate Pope, we implemented a new method to estimate the multidimensional SFS in an efficient way. Whilst it is still based on a likelihood function, it uses a binning heuristic approach alongside paired an accelerated expectation-maximization algorithm. This strategy allows for fast and memory-efficient results for both haploid and diploid genomes. The implementation supports folding and boostrapping procedures. We verified these expectations by means of extenstive simulations and by recovering previous findings of genetic differentiation in the fungal pathogen Neonectria neomacrospora.
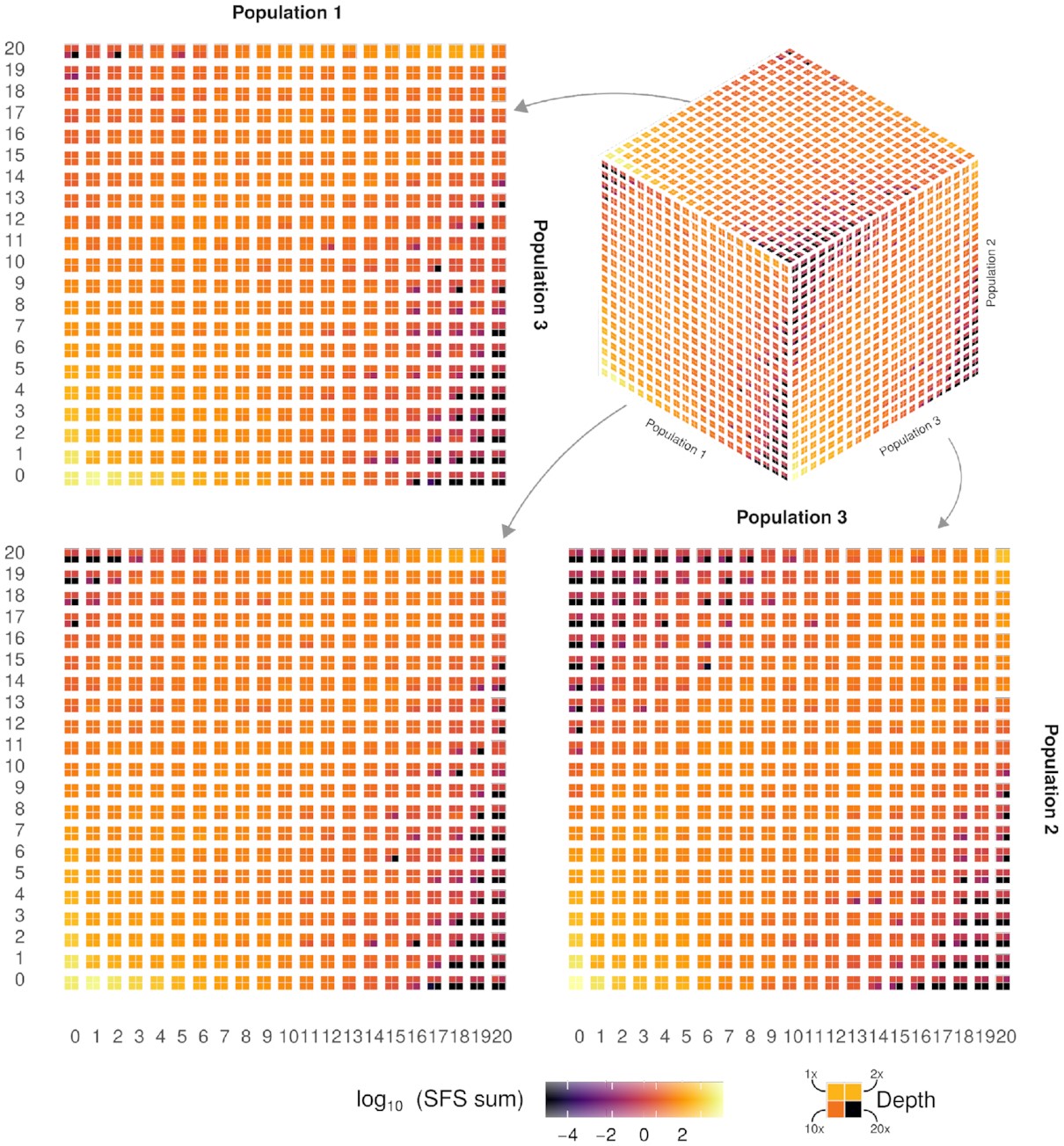
The new implementation opens up new possibilities of estimating the SFS for large and noisy data sets, whilst decreasing the experimental cost of generating high-quality genomes. It also spurs new new investigations for better optimisations of the likelihood function.
Return to our blog